
Um estudo recém publicado na Nature Medicine traz evidências de que vírus SARS CoV-2 surgiu a partir dos processos naturais de evolução dos seres vivos.
O novo coronavírus, identificado entre os cientistas como SARS-CoV-2 e que já teve seu genoma sequenciado por diversos estudos ao redor do mundo, passou recentemente por uma nova análise. Dessa vez os pesquisadores conduziram um estudo do genoma do vírus e o compararam com o genoma de outros vírus da família dos coronavírus. Esses pesquisadores acabam de publicar no importante periódico científico Nature Medicine o resultado dessa comparação, que permitiu a eles afirmarem que o SARS-CoV-2 não é produto de manipulação genética ou obtido em laboratório a partir de qualquer outro meio.
O resultado desse estudo, conduzido por pesquisadores dos Estados Unidos, Escócia e Austrália, põe fim à controvérsia sobre a origem do vírus SARS CoV-2 (também identificado como HCoV-19), que provoca a doença Covid-19 e é o responsável pela atual pandemia que já abrange mais de 70 países. O estudo, que foi descrito em carta publicada na revista Nature Medicine em 17 de março, traz evidências de que o SARS CoV-2 surgiu a partir dos processos naturais de evolução dos seres vivos. Os pesquisadores apontam mutações no genoma do vírus que o tornam mais infeccioso em seres humanos e que surgem aleatoriamente durante sua replicação. Essas variações genéticas são imperfeitas, o que torna improvável a hipótese de terem sido produzidas pelo homem.
“Ao comparar os dados disponíveis da sequência do genoma para cepas conhecidas de coronavírus, podemos determinar firmemente que o SARS-CoV-2 se originou através de processos naturais”, disse Kristian Andersen, que possui doutorado e é professor associado de imunologia e microbiologia da Scripps Research e autor correspondente no artigo.
“Os vírus têm genomas que não são muito grandes, então é possível sequenciá-los por inteiro de maneira bastante confiável, e estabelecer comparações entre as diversas sequências”, comenta ao do Jornal da USP o professor Daniel Lahr, do Departamento de Zoologia do Instituto de Biociências (IB) da USP e que não esteve envolvido no estudo. “O vírus SARS CoV-2, causador da Covid-19, tem um genoma com cerca de trinta mil pares de bases, enquanto o genoma humano tem aproximadamente três bilhões de pares de bases e a bactéria Escherichia coli, muito comum em experimentos laboratoriais, tem de 4 a 5 milhões pares de bases”. As bases são as unidades moleculares que formam o DNA, dispostas em pares na sua estrutura.
Além de Andersen, outros autores do artigo “A Origem Proximal do SARS-CoV-2” incluem Robert F. Garry, da Tulane University; Edward Holmes, da Universidade de Sydney; Andrew Rambaut, da Universidade de Edimburgo; e W. Ian Lipkin, da Universidade de Columbia. A pesquisa foi financiada pelos Institutos Nacionais de Saúde dos EUA, pela Pew Charitable Trusts, pelo Wellcome Trust, pelo Conselho Europeu de Pesquisa e pela ARC Australian Laureate Fellowship.
Os coronavírus são uma grande família de vírus que podem causar doenças que variam amplamente em gravidade. A primeira doença grave conhecida causada por um coronavírus surgiu com a epidemia de Síndrome Respiratória Aguda Grave (SARS) de 2003 na China. Um segundo surto de doença grave começou em 2012 na Arábia Saudita com a Síndrome Respiratória no Oriente Médio (MERS).
Em 31 de dezembro do ano passado, as autoridades chinesas alertaram a Organização Mundial de Saúde sobre um surto de uma nova cepa de coronavírus causando doença grave, que foi posteriormente denominada SARS-CoV-2. No dia 20 de fevereiro de 2020, eram quase 167.500 casos de COVID-19 foram documentados, embora muitos outros casos leves provavelmente não tenham sido diagnosticados. Chegando a 19 de março de 2020, o número de mortes causadas pelo vírus já somavam mais de nove mil pessoas, segundo um levantamento da universidade norte-americana Johns Hopkins.
Logo após o início da epidemia, os cientistas chineses sequenciaram o genoma do SARS-CoV-2 e disponibilizaram os dados para pesquisadores de todo o mundo. Os dados resultantes da sequência genômica mostraram que as autoridades chinesas detectaram rapidamente a epidemia e que o número de casos de COVID-19 aumentou por causa da transmissão de humano para humano após uma única introdução na população humana. Andersen e colaboradores de várias outras instituições de pesquisa usaram esses dados de sequenciamento para explorar as origens e a evolução do SARS-CoV-2, concentrando-se em vários recursos reveladores do vírus.
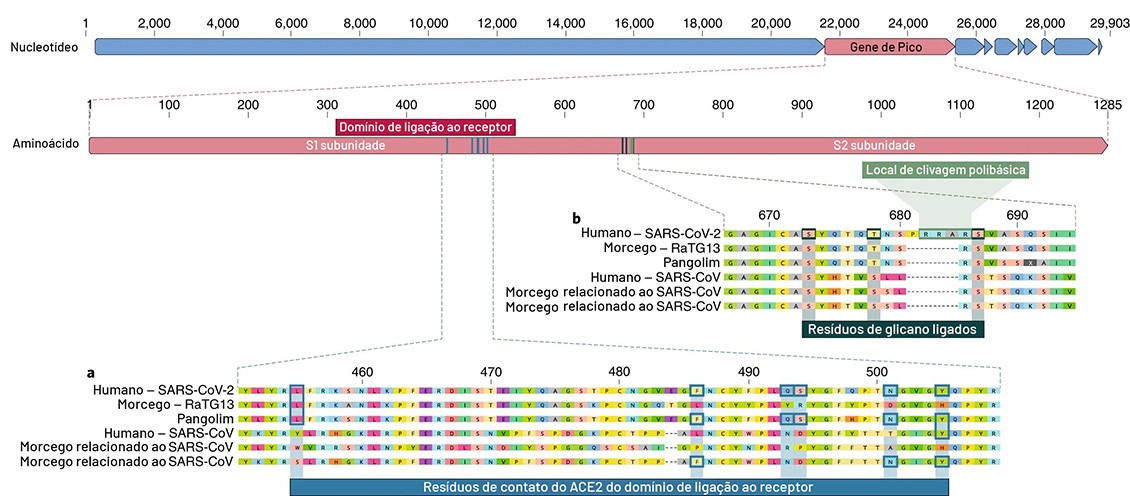
Os cientistas analisaram o modelo genético de proteínas SPIKE, armaduras do lado de fora do vírus que ele usa para agarrar e penetrar nas paredes externas das células humanas e de outros animais. Mais especificamente, os pesquisadores concentraram em duas características importantes da proteína SPIKE: o domínio de ligação ao receptor (RBD, sigla para “receptor-binding domain“), um tipo de gancho que agarra as células hospedeiras, e o local da clivagem, um abridor de latas molecular que permite que o vírus se abra e insira seu material genética nas células hospedeiras.
De acordo com Lahr, os pesquisadores, ao analisarem as variações de todo o genoma do vírus, conseguiram determinar que o SARS CoV-2 é muito proximamente relacionado com um vírus já descrito em morcegos, o RATG13. “Isso significa que eles possuem um hipotético ancestral comum”, destaca, “porém a observação de partes específicas do genoma indica semelhanças que, na comparação com outros vírus, dão a oportunidade de explicar eventos de evolução, e identificar as mutações mais importantes para infeccionar seres humanos”, explica o pesquisador ao repórter Júlio Bernardes do Jornal da USP. A matéria completa pode ser acessada neste link.
Evidências para evolução natural
A evidência para a evolução natural foi obtida a partir da análise dos dados mais fundamentais do SARS-CoV-2: sua estrutura molecular geral. Se alguém estivesse tentando projetar um novo coronavírus como patógeno, esse alguém teria construído esse novo patógeno a partir da genética de um vírus conhecido por causar doenças em humanos. Mas os cientistas descobriram que o material genético do SARS-CoV-2 diferia substancialmente de todos os coronavírus já conhecidos e que sua estrutura gênica se assemelhava principalmente com a de vírus da mesma família encontrados em morcegos e pangolins, um mamífero aparentado com o tatu encontrados na Ásia e na África .
“Essas duas características do vírus, as mutações na porção RBD da proteína SPIKE e seu genoma distinto, descartam qualquer hipótese de manipulação em laboratório como uma origem potencial para o SARS-CoV-2”, disse Andersen.
Josie Golding, PhD, líder de epidemias na Wellcome Trust, com sede no Reino Unido, disse que as descobertas de Andersen e demais pesquisadores autores do artigo são “de importância crucial para trazer uma visão baseada em evidências aos rumores que circulam sobre as origens do vírus (SARS-CoV -2) causando COVID-19”. Goulding ainda acrescenta que “os pesquisadores concluem que o vírus é o produto da evolução natural, encerrando qualquer especulação sobre engenharia genética deliberada”.
Possíveis origens do vírus
Com base em sua análise de sequenciamento genômico, Andersen e seus colaboradores concluíram que as origens mais prováveis para o SARS-CoV-2 seguiram um de dois cenários possíveis.
Em um primeiro cenário, o vírus evoluiu para seu estado patogênico atual por meio da seleção natural em um hospedeiro não humano e depois passou a infectar os seres humanos. Foi assim que surgiram os surtos anteriores de coronavírus, com humanos contraindo o vírus após exposição direta a civetas, um animal pequeno classificado m uma família de mamíferos carnívoros e camelos, como se deu para a SARS e a MERS, respectivamente. Os pesquisadores propuseram os morcegos como o reservatório natural mais provável para o SARS-CoV-2, já que esse vírus é muito semelhante a um coronavírus conhecido por infectar morcegos. No entanto, não há casos documentados de transmissão direta de morcego para humano, sugerindo aos cientistas que um hospedeiro intermediário provavelmente esteja envolvido no caminho que o vírus fez de morcegos até chegar a humanos.
Em um outro cenário proposto, uma versão não patogênica do vírus saltou de um animal hospedeiro direto para o ser humano e depois evoluiu para seu estado patogênico atual na população humana. Por exemplo, alguns coronavírus de pangolins têm uma estrutura RBD muito semelhante à do SARS-CoV-2. É possível que um coronavírus de um pangolim possa ter sido transmitido a um ser humano, diretamente ou através de um hospedeiro intermediário, como as civetas, o animal criado muitas vezes sob condições péssimas para produzir o café mais caro do mundo, o Kopi Luwak, ou café de civeta.
Então, a outra característica distinta da proteína SPIKE do SARS-CoV-2, o local de clivagem, poderia ter evoluído dentro de um hospedeiro humano, possivelmente através da circulação entre humanos não detectada antes do início da epidemia. Os pesquisadores descobriram que o local de clivagem SARS-CoV-2 é semelhante aos locais de clivagem de cepas de gripe aviária, para as quais já foi demonstrado que são facilmente transmitida para as pessoas. O SARS-CoV-2 poderia ter desenvolvido um local de clivagem tão virulento nas células humanas e logo depois viria a desencadear a epidemia atual, à medida que o coronavírus provavelmente teria se tornado muito mais capaz de se espalhar entre humanos.
O co-autor do estudo, Andrew Rambaut, alertou, segundo o site Science Daily, que é difícil, senão impossível, saber neste momento qual dos cenários é o mais provável. Mas se o SARS-CoV-2 infectou os seres humanos em sua forma patogênica atual a partir de uma fonte animal, aumenta a probabilidade de futuros surtos, pois a cepa do vírus causadora de doenças ainda pode estar circulando na população animal e pode novamente saltar para humanos em novas formas disparar novas epidemias com a que vivenciamos agora. Porém, as chances disso acontecer são menores se um coronavírus não patogênico entrou na população humana e depois desenvolveu as propriedades patogênicas do SARS-CoV-2.





{kind=link}